
I recently wrote about workflow management systems in bioinformatics, focusing on Nextflow and Snakemake. In this post, the aim is to compose a small Bioinformatics workflow to start exploring the Nextflow syntax and its features. The Nextflow documentation is extensive and provides many examples that are a helpful start. Nevertheless, it is still challenging to go from the snippets provided to developing your own workflows.
Basic concepts
Nextflow is a reactive workflow framework and a programming DSL designed for writing data-intensive computational pipelines. Nextflow adds the ability to define complex program interactions and a high-level parallel computational environment based on the dataflow programming model.
Workflows are composed by joining together different processes. These define the basic processing units of the work that need to be executed and can be written in a variety of scripting languages (Bash, Python, etc.). Processes typically are executed as a single task or, if appropriate, executed as multiple tasks, as many times as required in parallel (e.g. processing multiple files).
Processes are isolated from each other, and the only communication between them is through channels.
Any process can define one or more channels as input or output.
Unless explicitly declared with the DSL 2 workflow definition workflow {...},
the workflow is implicitly defined by these input and output declarations.
Bioinformatics workflow
Developing a bioinformatics workflow with Nextflow presents a good opportunity to explore Nextflow and see some its features in action. The workflow we are developing here is very simple but a good place to start.
- Download SwissProt sequence dataset
- Generate a SwissProt NCBI Blast database
- Perform a sequence search with a sequence of interest against the SwissProt database
- Extract the complete sequences of the BLAST top hits
The first consideration to make is whether we should go for Nextflow DSL 2 or default to DSL 1.
Looking forward, the likelihood is that DSL version 2 will be the default and therefore, in this example, we should
set nextflow.enable.dsl=2.
In this example, we need to run a few of the BLAST commands. For that, we can install blast locally with Conda,
simply by running conda install -c bioconda blast==2.12.0.
In the Nextflow script file, we can include paths to these
tools, so we can use them in the processes later on. We should also add other parameters,
which makes changing them easier later on, rather than having them hard coded.
The beginning of our workflow_example.nf script should look like this:
// Declare syntax version
nextflow.enable.dsl=2
// Script parameters
// BLAST commands
params.makeblastdb = "/Users/username/miniconda3/envs/py39/bin/makeblastdb"
params.blastp = "/Users/username/miniconda3/envs/py39/bin/blastp"
params.blastdbcmd = "/Users/username/miniconda3/envs/py39/bin/blastdbcmd"
// SwissProt EBI FTP URL
params.ftp_url = \
"https://ftp.ebi.ac.uk/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz"
// Work directory
params.workdir = "/Users/username/tmp/"
// Query sequence
params.input_fasta = "/Users/username/tmp/query.fasta"
// (optional) SwissProt database name
params.dbname = "swissprot"Now we need to start adding the workflow processes to the workflow_example.nf file.
The first process 1 is about downloading the SwissProt dataset. For that we need to specify the
expected output path, which we can define as "swissprot.fasta", for example.
Since we need this dataset in the next process, we can emit it, explicitly as swissprot, or we could
simply use "swissprot.fasta" as an expected input of the next process. The script component of the process
simply is taking care of downloading the compressed dataset and then uncompressing it.
// downloads the SwissProt dataset from the EBI's FTP
process downloadSwissProt {
output:
path "swissprot.fasta", emit: swissprot
script:
"""
wget ${params.ftp_url} -O swissprot.fasta.gz
gzip -d swissprot.fasta.gz
"""
}In the next process 2, we take the SwissProt dataset and generate a BLAST database. We take the emitted file and
expect several files to be generated by the makeblastdb command. Note how we are using the parameters in the
script part of the process. Here, we are explicitly emitting the swissprotdb files.
// Builds the SwissProt BLAST database
process makeBlastDb {
input:
path swissprot
output:
path "swissprot*", emit: swissprotdb
script:
"""
${params.makeblastdb} -dbtype 'prot' \
-in ${swissprot} -out ${params.dbname} \
-input_type 'fasta' -blastdb_version 5 -parse_seqids
"""
}In the next process 3, we take the BLAST database and a query sequence of interest and perform sequence searching. We then process the BLAST output, and generate a temporary list of the 10 top hits (i.e. the hits with smaller e-values, or hits believed to be the closest homologs in the database).
// Performs a BLASTP sequence search and captures the 10 top Hits
process blastSearch {
input:
path query
path swissprotdb
output:
path "top_hits.txt"
script:
"""
${params.blastp} -db ${params.dbname} -query ${query} -outfmt 6 > blast_result
cat blast_result | head -n 10 | cut -f 2 > top_hits.txt
"""
}Finally, in 4, we take those 10 hits and extract the complete sequences in fasta format using the blastdbcmd
command. Note the addition of publishDir params.workdir as we want the top_hits.fasta file to be available
in our work directory, so we can continue working on the hit sequences. The same can be done on any of the
other processes, for example, the makeBlastDb process, where we generate the SwissProt BLAST database, and
might want to store it more permanently outside the Nextflow work directories.
// Extracts top Hit sequences in fasta format
process extractTopHits {
publishDir params.workdir
input:
path top_hits
path swissprotdb
output:
path "top_hits.fasta"
script:
"""
${params.blastdbcmd} -db ${params.dbname} -entry_batch ${top_hits} > top_hits.fasta
"""
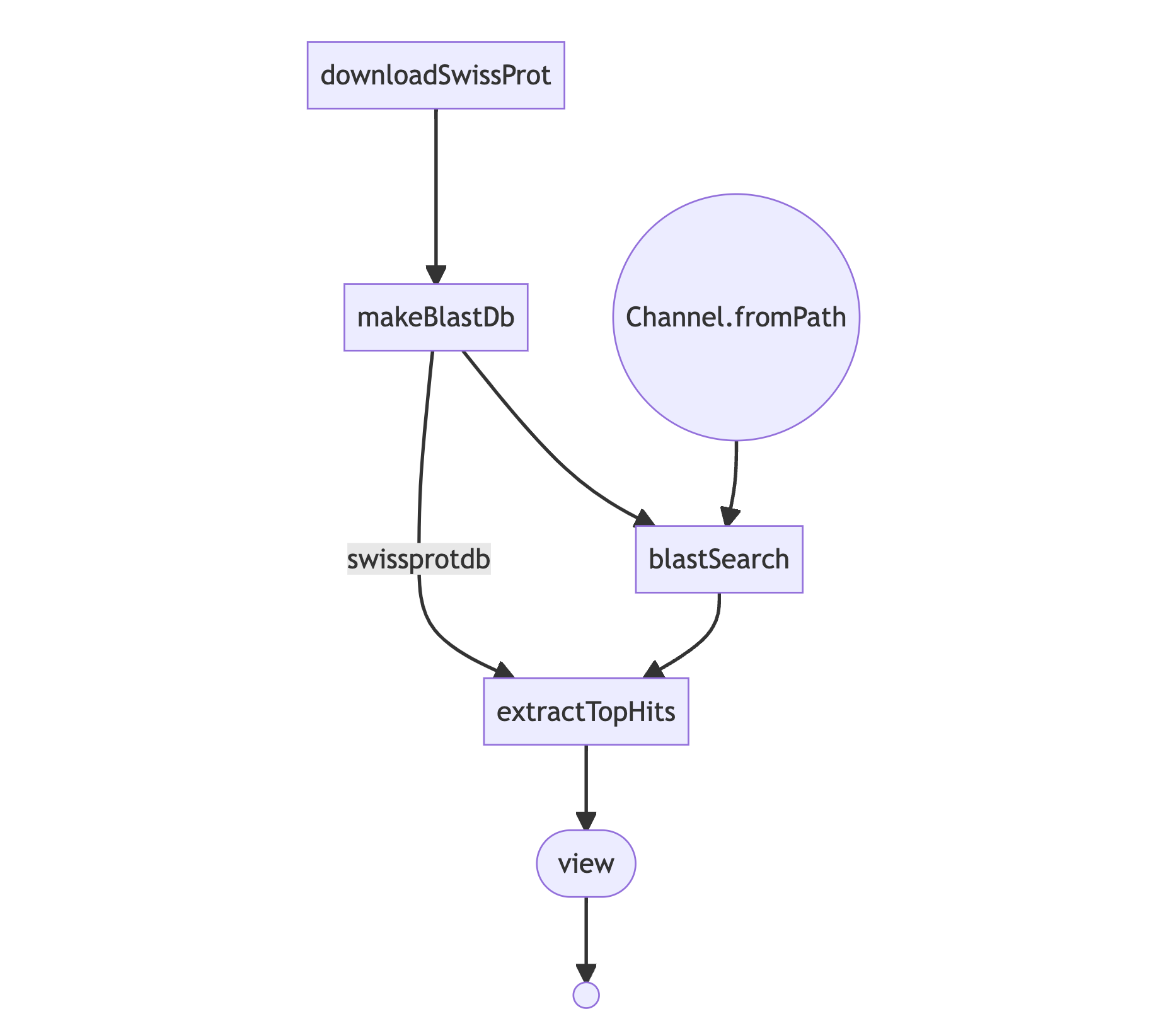
}The processes are defined, and now we need to define the workflow. With DSL 2, we can use workflow, which, for this
example, could be as shown below. Note that blastSearch and extractTopHits are using the outputs from the
previous processes, which is the same for makeBlastDb. The difference in the latter is that we are simply
piping (i.e. chaining) the commands together.
// workflow definition
workflow {
downloadSwissProt() | makeBlastDb
def query_ch = Channel.fromPath(params.input_fasta)
blastSearch(query_ch, makeBlastDb.out)
extractTopHits(blastSearch.out, makeBlastDb.out) | view
}The workflow_example.nf file is now complete and ready to use. We could simply run
nextflow run example_worflow.nf and use the -resume flag to re-run it if we modified some processes.
Nextflow has functions to generate DAG graphs (as exampled below), but also to produce several reports,
for resource usage, tasks, timeline, trace report, etc.
The beauty of Nextflow is that we can execute this locally in our own machine, but likewise we can run it on a
high-performance computing (HPC), without much modification.
For example, we could set a couple of profiles as shown below and simply call
nextflow run example_worflow.nf -profile hpc, passing the profile we want from the config.
profiles {
default {
process.executor = 'local'
}
hpc {
process.executor = 'lsf'
process.queue = 'standard'
}
}For that we need to save the profiles config as .nextflow/config on our work directory.
Alternatively, we could add executor and clusterOptions directive declarations to the processes as required.
For example:
// Performs a BLASTP sequence search and captures the 10 top Hits
process blastSearch {
executor 'lsf'
queue 'standard'
input:
path query
path swissprotdb
output:
path "top_hits.txt"
script:
"""
(...)
"""
}In addition to these, there are a plethora of other directives we could use, for example, to specify
CPU other requirements: cpus , memory and disk.
There are directives afterScript and beforeScript scripts,
for retrying and errorStrategy, for caching, for Conda and containers, and the list goes on.
This is just touching the surface of what we can do with Nextflow. Let me know if this was useful! Share your experiences and your thoughts about Nextflow!