
Several alternative scientific workflow systems typically referred to as workflow management platforms are used to run data pipelines (e.g. SNP calling and performing ETL). Among the most popular workflow managers are Nextflow and Snakemake. These were conceived in Bioinformatics labs but essentially try to address similar reproducibility and scalability issues that other general-purpose systems try to solve, for example, Apache Airflow and Luigi.
Both Nextflow and Snakemake were launched almost 10 years ago, stemming from the need for processing increasing quantities of scientific data, namely Next-Generation Sequencing (NGS) data. These projects provide extensive documentation and examples. Both systems promise easy learning and adoption, as well as workflow reproducibility and scalability. From their homepages, one can read:
Nextflow enables scalable and reproducible scientific workflows using software containers. It allows the adaptation of pipelines written in the most common scripting languages. Its fluent DSL simplifies the implementation and the deployment of complex parallel and reactive workflows on clouds and clusters.
…
Snakemake (…) is a tool to create reproducible and scalable data analyses. Workflows are described via a human readable, Python based language. They can be seamlessly scaled to server, cluster, grid and cloud environments, without the need to modify the workflow definition. Finally, Snakemake workflows can entail a description of required software, which will be automatically deployed to any execution environment.
…
The approach taken by both systems is to break down the pipelines into tasks or processes and link them into workflows through inputs and outputs. Tasks or processes are executed when inputs are received, but both inputs and outputs typically need to be defined explicitly for the workflows to be validated and executed.
The systems use custom domain-specific language (DSL) extensions of Groovy and Python, for Nextflow and Snakemake, respectively. The popularity of Python among the Bioinformatics community would suggest Snakemake would be the most adopted option, but it turns out that Nextflow is very popular and the choice of many researchers and groups. A benefit in both cases is the ability to use the underlying scripting languages beyond the DSL as required. The composed workflows can be visualised as DAGs (direct acyclic graphs), and both systems provide functions to generate them. See an example of a Nextflow DAG below.
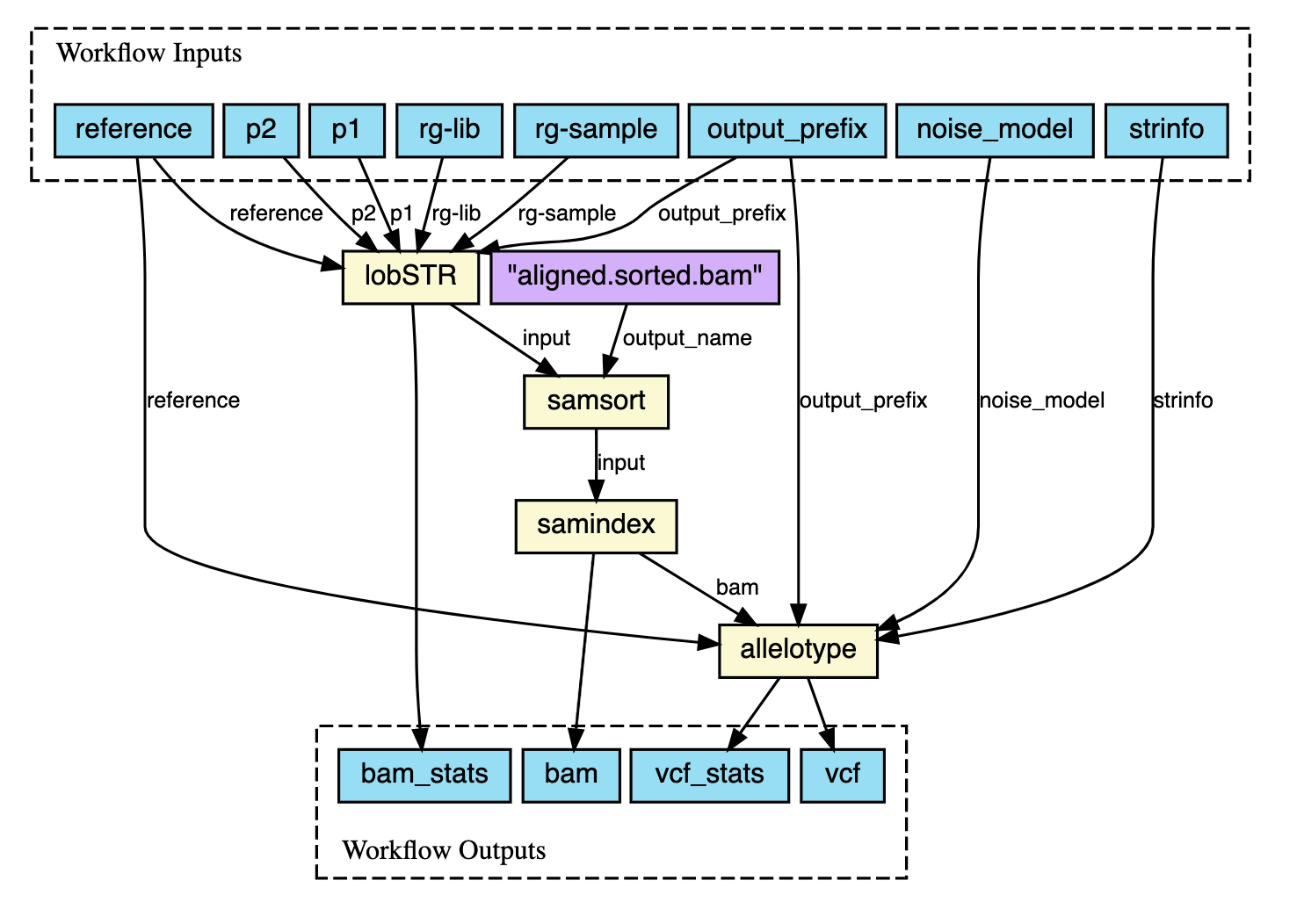
Both systems enable the automatic parallelization of jobs and enable automatic retrying of failed jobs. Nextflow provides
high-performance computing (HPC) executors out of the box, supporting traditional HPC,
running Slurm, IBM LSF, SGE and others, but also supports execution on public cloud providers,
such as Amazon AWS, Google Cloud and Microsoft Azure. Snakemake can similarly be executed in such environments
via profiles. Additionally, both workflow management systems support cloud execution via Kubernetes.
Both systems also enable the portability of the workflows by supporting execution
on different computing environments (e.g. Conda environments) and container technology
(e.g. Docker, Singularity, etc.).
Snakemake allows dry-run execution testing, which the Nextlow command-line interface
(CLI) only supports on selected commands (e.g. nextflow clean ...)
These workflow systems provide a great way for researchers to make reproducible data pipelines. The nf-core community effort, for instance, provides a curated set of analysis pipelines built with Nextflow that anyone can re-use.
Nextflow and Snakemake constitute solid choices for developing scientific workflows. Some particular features might be what makes you pick one of them over the other, but overall either are very capable. One could use both systems side by side if that fits a particular user requirement.
I am not going to declare a winner or pick a favourite, but do let me know over on Twitter which you use and why!